
A Two-Hit Model of The Biological Origin of Posttraumatic Stress Disorder (PTSD)
Apostolos P. Georgopoulos1,2,3,4,5*, Lisa M. James1,2,3,4, Peka Christova1,2,3, Brian E. Engdahl1,2,3,6
1 Brain Sciences Center, Department of Veterans Affairs Health Care System, Minneapolis, Minnesota, USA
2 Department of Neuroscience, University of Minnesota Medical School, Minneapolis, Minnesota, USA
3 Center for Cognitive Sciences, University of Minnesota, Minneapolis, Minnesota, USA
4 Department of Psychiatry, University of Minnesota Medical School, Minneapolis, Minnesota, USA
5 Department of Neurology, University of Minnesota Medical School, Minneapolis, Minnesota, USA
6 Department of Psychology, University of Minnesota Medical School, Minneapolis, Minnesota, USA
Abstract
Posttraumatic stress disorder (PTSD) is a debilitating disorder that can develop following exposure to a traumatic event. Although the cause of PTSD is known, the brain mechanisms of its development remain unknown, especially why it arises in some people but not in others. Most of the research on PTSD has dealt with psychological and brain mechanisms underlying its symptomatology, including intrusive memories, fear and avoidance (see ref.1 for a broad coverage of PTSD research)1. Here we focus, instead, on the origin of PTSD, namely on the neural mechanisms underlying its development. Specifically, we propose a two-hit model for PTSD development, with the following components. (a) The 1st hit is a neuroimmune challenge, as a preexisting condition, and the 2nd hit is intense glutamatergic neurotransmission, induced by the traumatic event; (b) the key molecule that mediates the effects of these two hits is intercellular adhesion molecule 5 (ICAM-5) which was found to be differentially expressed in PTSD2. ICAM-5 is activated by neuroimmune challenge3,4 and glutamatergic neurotransmission5,6, it further enhances glutamatergic transmission6, and exerts a potent effect on synapse formation and neural plasticity, in addition to immunoregulatory functions3,4,7; and (c) with respect to the neural network(s) involved, the brain areas most involved are medial temporal cortical areas, and interconnected cortical and subcortical areas8-10. We hypothesize that the net result of intense glutamatergic transmission in those areas induced by a traumatic event in the presence of ongoing neuroimmune challenge leads to increased levels of ICAM-5 which further enhances glutamatergic transmission and thus leads to a state of a neural network with highly correlated neural interactions, as has been observed in functional neuroimaging studies8-10. We assume that such a “locked-in” network underlies the intrusive re-experiencing in PTSD and maintains associated symptomatology, such as fear and avoidance.
Introduction
Posttraumatic stress disorder (PTSD) is a psychiatric disorder that can develop in response to exposure to traumatic events, with hallmark symptoms including intrusive recollections or re-experiencing of the traumatic event, avoidance of trauma reminders, cognitive and affective changes, and hyperarousal11. PTSD is associated with high rates of psychiatric and medical comorbidities12-16, and increased mortality17,18. Although 90% of individuals are exposed to potentially traumatic events, only 7-10% of the general population19,20 and 10-30% of veterans21-23, develop PTSD suggesting that trauma alone is not sufficient. Rather, the typical outcome following trauma exposure is resilience24. Nonetheless, for those who develop PTSD, the symptoms may lead to significant chronic functional impairment. Furthermore, although several psychological and pharmacological interventions have been shown to be effective in reducing symptoms, treatment outcomes vary considerably with many individuals failing to respond and up to two-thirds of treatment responders failing to achieve remission25, leading many to emphasize the need for novel interventions.
Current Theories of PTSD
Numerous psychological and neurobiological theories of PTSD have been proposed. Psychological theories generally focus on cognitive functions associated with trauma including abnormal information processing, maladaptive interpretations about traumatic experiences, deleterious effects of trauma on belief systems, and memory disturbances, as well as affective contributions to these processes (several dominant models are reviewed in ref.26). Similarly, several neurobiological theories of PTSD have emerged and are summarized elsewhere27. Briefly, the dominant models focus on dysfunctional fear learning, threat detection, executive function/emotion regulation, and contextual processing and the associated neural regions including the amygdala, midcingulate cortex, insula, medial prefrontal cortex, and the hippocampus. These theories have been useful in explaining PTSD-associated symptomatology and have, in some cases, provided the basis for psychological interventions; however, they largely fail to adequately explain the development of PTSD. Thus, here we present a novel model of PTSD that elucidates the origin of the disorder, accounts for resilient vs pathological trajectories following trauma exposure, and provides an explanation for treatment non-response.
Biological Correlates of PTSD
Biological correlates of PTSD have been widely investigated with numerous studies identifying physiological, neuroendocrine, and genetic factors in addition to structural and functional brain alterations (for reviews see refs.28,29). At the brain network level, anomalous neural interactions characterized by hypercorrelation9 have been shown to distinguish PTSD from controls8,10, with both cortical9 and limbic regions involved30. Based on those findings, we hypothesized that the persistence of highly correlated neural ensembles underlies characteristic PTSD symptoms30. Here we discuss the role of immune system involvement in facilitating the persistent hypercorrelated network associated with PTSD.
Immunological involvement in PTSD is a promising avenue that has recently emerged. Recent reviews29,31 note immune system alterations including increased levels of pro-inflammatory cytokines, C-reactive protein (CRP), and tumor necrosis factor (TNF)-α in PTSD. Notably, cytokines have been found to have profound effects on the HPA axis32, which is intimately linked to PTSD. In addition, FKBP5, a gene involved in immune regulation and a modulator of HPA axis function have been implicated in PTSD33. One particularly noteworthy study demonstrated that pre-deployment CRP predicted post-deployment PTSD34, suggesting inflammation may be a predisposing factor for PTSD. To that end, it has been speculated that inflammation resulting from traumatic brain injury underlies the high rates of co-occurrence of brain injury and PTSD35. Finally, changes in inflammatory markers have been reported following PTSD treatment31, and administration of hydrocortisone, an anti-inflammatory agent, immediately after trauma exposure has been shown to prevent PTSD in human subjects and promote dendrite growth and spine density in stressed animals36. In fact, those latter positive neural changes extended to other domains as well, including increased levels of brain-derived neurotrophic factor. These findings underscore the intricate interplay among neural, hormonal and inflammatory factors in serious stress and the possible alleviation of its deleterious effects.
Biological Basis of the Origin of PTSD: A Two-Hit Model
PTSD is typically initiated by massive multi-sensory activation produced by the trauma exposure30. The layout of cortical connectivity, as delineated by Jones and Powell37, channels the flow of activation from primary cortical sensory areas, through intermediate interconnected stations, to ultimate convergence in the parieto-temporal and frontal cortices, and therefrom distribution to rostral and medial temporal areas, anterior and orbitofrontal frontal areas, and subcortical areas38. We have proposed that these multiple and converging interactions induce stable correlations among the various areas of convergence30, an idea supported by the results of modeling studies30. Such stable correlations have been observed in PTSD9, in contrast to healthy controls where a systematic reduction in trauma-related neural correlations was observed9. We have hypothesized that this stable correlation pattern represents a “locked-in” state of the late-stage sensory processing that underlies the occurrence of intrusive traumatic re-experiences and flashbacks8. It is reasonable to suppose that trauma exposure will initially induce such correlations to all brains; the question then is: why such correlations would persist in PTSD with all the untoward consequences? The considerations above and the findings of recent studies led us to propose a two-hit model for PTSD development, with the following components. (i) The 1st hit is neuroimmune challenge, as a preexisting condition; (ii) the 2nd hit is intense glutamatergic neurotransmission, induced by the traumatic event; (iii) the key molecule that mediates the effects of these two hits is intercellular adhesion molecule 5 (ICAM-5) which was found to be differentially expressed in PTSD2 (it is very possible that other molecules, in addition to ICAM-5, might be involved in this process); and (iv) the net result is a hypercorrelated state among higher-order association cortical and subcortical areas, with a focus on the temporal lobe8-10. Specifically, we hypothesize that the net result of intense glutamatergic transmission in those areas induced by a traumatic event in the presence of ongoing neuroimmune challenge leads to increased levels of ICAM-5 which further enhances glutamatergic transmission and thus leads to a state of a neural network with highly correlated neural interactions, as has been observed in functional neuroimaging studies8-10. We assume that such a “locked-in” network underlies the intrusive re-experiencing in PTSD and maintains associated symptomatology, such as fear and avoidance.
The Role of InterCellular Adhesion Molecule 5 (ICAM-5)
A recent study using transcriptomic analysis immediately following exposure to a traumatic event found that ICAM-5 was differentially expressed in people who subsequently developed PTSD following that exposure2. This finding plays a central role in our model, based on the dual role of this molecule, namely in glutamatergic neurotransmission5,6 and local immunosuppression3,4. ICAM-5 is a member of the immunoglobulin superfamily, a group of adhesive glycoproteins expressed on cell surfaces that are primarily involved in cellular processes underlying the immune system and inflammation. ICAM-5, however, is unique in that it is the only ICAM that is expressed exclusively in the brain39, and it has been shown to play a critical role in dendritic development, synapse maturation, and associated neural network plasticity, in addition to typical ICAM immunoregulatory functions3-7. Moreover, ICAM-5 expression is limited to neurons in select telencephalic regions, including the neocortex, hippocampus, amygdala, caudate nucleus, and putamen, areas that are involved in higher-order functions such as learning and memory. For biological action, ICAM-5 is cleaved resulting in soluble ICAM-5 (sICAM-5); sICAM-5 enhances glutamatergic neurotransmission6 and exerts local immunosuppression relating to both innate and adaptive immunity3,4. Both of these actions bear directly on our hypothesis regarding its role in inducing and maintaining PTSD’s intrusive memory and traumatic re-experiencing symptomatology.
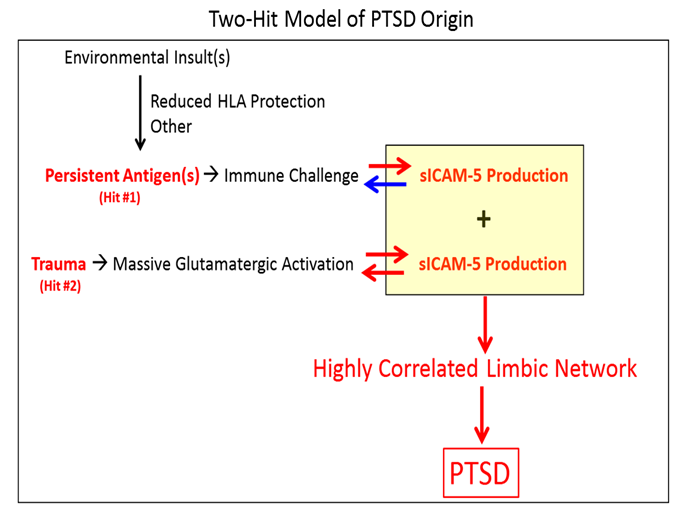
Figure 1: Schematic diagram of the two-hit model of PTSD origin. Red arrows indicate enhancing effect; blue arrow indicates immunosuppressive effect. See text for details.
ICAM-5 exerts an immunosuppressive action, as mentioned above, in response to antigenic challenges. sICAM-5 is released in response to viral infections, including encephalitis of diverse causes40. Its role helps keep local inflammation at low levels. Therefore, it is reasonable to suppose that sICAM-5 will be produced in response to local antigenic challenges. According to our model, it is the occurrence of a traumatic event in such a background that is crucial for PTSD development.
Glutamatergic Neurotransmission Enhances sICAM-5 production, and Vice Versa
In the realm of synaptic transmission, independently of antigenic challenge, the key factor producing ICAM-5 cleavage, and hence production of sICAM-5, is activation of glutamate receptors, specifically N-methyl-D-aspartate (NMDA)5 and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors6. sICAM-5, in turn, enhances glutamatergic transmission6, thus creating a vicious cycle of an activated network. Since the dominant, if not exclusive, excitatory neurotransmission in the cerebral cortex is glutamatergic, it follows that the massive neuronal activity initiated by a traumatic event will lead to increased levels of sICAM-5, which, in turn, will further enhance glutamatergic transmission.
We hypothesize that this results in “fixing” those synapses, which, at the network level, would represent the memorized experience of the traumatic event. If that were universally the case for all people, why does PTSD develop in some but not others? For this, we postulate that the local level/concentration of sICAM-5 is the crucial factor: the higher the sICAM-5 level, the stronger the synaptic “tightness”, and the associated correlations. This would explain why more serious traumatic events would be likely to have more lasting consequences but could not explain how traumatic events of similar intensity would lead to PTSD in some but not others. For this, we postulate a second hit, as follows.
PTSD: Intense Glutamatergic Transmission in The Background of Immune Challenge
We postulate that intense glutamate transmission induced by a major traumatic event occurring in the background of ongoing antigenic/immunological challenge will lead to a large increase of sICAM-5 leading, in turn, to substantially enhanced synaptic maturation. We further postulate that this essentially “fixes” the synaptic landscape of the network activated by the traumatic event. As mentioned above, this network mainly comprises anterior and medial temporal areas, anterior frontal and orbitofrontal areas, and subcortical limbic areas8-10. In this context, we explain the observed relation between ICAM-5 production and PTSD development2 by hypothesizing that ICAM-5 production was a pre-existing condition due to inflammatory/immunogenic challenge on the background of which the massive glutamate input induced a synaptic “locking” of the activated network, manifested behaviorally as persistent re-experiencing of the traumatic event. This interpretation is consistent with the findings of 3 studies, namely (a) that an elevated blood CRP is predictive of PTSD development34, (b) that intravenous (IV) injection of hydrocortisone (an anti-inflammatory agent) shortly after the occurrence of the traumatic event reduced the chance of PTSD development36, and (c) that IV injection of ketamine (a noncompetitive NMDA antagonist with relatively selective action in the midcingulate cortex41) in chronic PTSD alleviates the severity of its symptoms42. This last study, in particular, is in keeping with our hypothesis that a persisting, enhanced glutamatergic transmission underlies PTSD symptomatology.
Background Immune Challenge: Persistent Antigens and PTSD
Our two-hit model for PTSD development predicts that exposure to trauma in the presence of neuroimmune challenge would increase their risk of developing PTSD. As mentioned above, the presence of inflammation, as indicated by increased plasma CRP, is a risk factor for developing PTSD following trauma exposure34 and, conversely, the presence of PTSD is a risk factor for subsequently developing an autoimmune disease43,44. Moreover, we found in a recent study45 that Gulf War Illness (GWI), an immune-related disorder46-48, primes the occurrence of PTSD.
The various mechanisms underlying the interplay between PTSD and associated immune-related comorbidities, and the relevant neuroendocrine alterations involved, have been discussed succinctly49. In general, it is not clear which is the primary cause. It is very possible that PTSD and immune-related disorders share common functional alterations, predisposing for each other, that will need further research to be disentangled. Based on our initial findings of reduced Human Leukocyte Antigen (HLA) class 2 protection in GWI46, and on our more recent finding that HLA allele DRB1*13:02 prevents brain atrophy in GWI48, we proposed48 that an important underlying factor for GWI consists of “persistent antigens”, that is, antigens (of various kinds) to which veterans of the 1991 Gulf War were exposed (most probably through the vaccines received) but which could not be eliminated due the lack of HLA protection46. This hypothesis is in keeping with the results of our study51 that blood serum from veterans suffering from GWI exerts detrimental effects on brain cultures that are prevented by the addition of serum from healthy Gulf War era veterans. Finally, the link between the “persistent antigen” hypothesis47 and immune-related disorders is reinforced by the protection conferred by DRB1*13:02 on both GWI47 and a broad range of typical autoimmune diseases50. Thus, the underlying immune challenge we propose in our two-hit PTSD model here could be attributed to persistent antigens that would reflect reduced adaptive immunity protection in susceptive individuals. This issue remains to be clarified by an extensive investigation of HLA makeup in PTSD. In addition, the nature of the hypothesized persistent antigens could be investigated by experiments employing neural cultures51. Using such cultures, we have shown that deleterious effects of persistent antigens circulating in the blood can be reduced or eliminated51; this opens the possibility of treating PTSD by identifying and eliminating persistent antigens that otherwise hinder neural plasticity and recovery.
Acknowledgments
This study was funded by the University of Minnesota (the William L. Anderson Chair in PTSD Research and the American Legion Brain Sciences Chair) and the U.S. Department of Veterans Affairs. The sponsors had no role in the writing of this paper. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
- Georgopoulos AP, James LM, Engdahl BE. Neural mechanisms of posttraumatic stress disorder. Frontiers Research Topic 2017. https://www.frontiersin.org/research-topics/3447/neural-mechanisms-of-posttraumatic-stress-disorder.
- Wingo A, Daskalakis N, Galatzer-Levy I, et al. Transcriptome-Wide Analysis identifies ICAM5 differentially expressed in chronic PTSD symptoms versus resiliency post trauma exposure in a longitudinal study. Biol Psychiatry. 2018; 83: 91-92.
- Tian L, Lappalainen J, Autero M, et al. Shedded neuronal ICAM-5 suppresses T-cell activation. Blood. 2008; 111: 3615-3625.
- Tian L, Rauvala H, Gahmberg CG. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009; 30: 91-99.
- Tian L, Stefanidakis M, Ning L, et al. Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J Cell Biol. 2007; 178: 687-700.
- Lonskaya I, Partridge J, Lalchandani RR, et al. Soluble ICAM-5, a product of activity dependent proteolysis, increases mEPSC frequency and dendritic expression of GluA1. PLoS One. 2013; 8: e69136.
- Ning L, Tian L, Smirnov S, et al. Interactions between ICAM-5 and β1 integrins regulate neuronal synapse formation. J Cell Sci. 2013; 126: 77-89.
- Engdahl BE, Leuthold AC, Tan H-RM, et al. Post-traumatic stress disorder: A right temporal lobe syndrome. J Neural Eng. 2010; 7: 066005.
- James LM, Engdahl BE, Leuthold AC, et al. Neural network modulation by trauma as a marker of resilience: differences between veterans with posttraumatic stress disorder and resilient controls. JAMA Psychiatry. 2012; 70: 410-418.
- Christova P, James LM, Engdahl BE, et al. Diagnosis of posttraumatic stress disorder (PTSD) based on correlations of prewhitened fMRI data: outcomes and areas involved. Exp Brain Res. 2015; 233: 2695-2705.
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5. Arlington, VA: Author; 2013.
- Boscarino JA. Posttraumatic stress disorder and physical illness: results from clinical and epidemiologic studies. Ann N Y Acad Sci. 2004; 1032: 141–153.
- Kubzansky LD, Koenen KC. Is posttraumatic stress disorder related to development of heart disease? An update. Cleve Clin J Med. 2009; 76(Suppl 2): 60–65.
- Nazarian D, Kimerling R, Frayne SM. Posttraumatic stress disorder, substance use disorders, and medical comorbidity among returning U.S. veterans. J Trauma Stress. 2012; 25: 220-5.
- O’Donovan A, Cohen BE, Seal KH, et al. Elevated risk for autoimmune disorders in Iraq and Afghanistan veterans with posttraumatic stress disorder. Biol Psychiat. 2015; 77: 365–374.
- Pietzrak RH, Goldstein RB, Southwick SM, et al. Prevalence and Axis I comorbidity of full and partial posttraumatic stress disorder in the United States: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. J Anxiety Disord. 2011; 25: 456-465.
- Boscarino JA. Posttraumatic stress disorder and mortality among US Army veterans 30 years after military service. Annals Epidemiol. 2006; 16: 248-256.
- Schlenger WE, Corry NH, Williams CS, et al. A prospective study of mortality and trauma-related risk factors among a nationally representative sample of Vietnam veterans. Am J Epidemiol. 2015; 182: 980-990.
- Kessler RC, Sonnega A, Bromet E, et al. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiat. 1995; 52: 1048-1060.
- Kilpatrick DG, Resnick HS, Milanak ME, et al. National estimates of exposure to traumatic events and PTSD prevalence using DSM-IV and DSM-5 criteria. J Trauma Stress. 2013; 26: 537-547.
- Kang HK, Natelson BH, Mahan CM, et al. Post-Traumatic Stress Disorder and Chronic Fatigue Syndrome-like illness among Gulf War Veterans: A population-based survey of 30,000 Veterans. Am J Epidemiol. 2003; 157: 141-148.
- Tanielian T, Jaycox, L. (Eds). Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. Santa Monica, CA: RAND Corporation, 2008.
- Weiss DS, Marmar CR, Schlenger WE, et al. The prevalence of lifetime and partial post?traumatic stress disorder in Vietnam theater veterans. J Trauma Stress. 1992; 5: 365-376.
- Marmar CR, Shlenger W, Henn-Haase C, et al. Course of posttraumatic stress disorder 40 years after the VietnamWar: findings from the National Vietnam Veterans Longitudinal Study. JAMA Psychiatry. 2015; 72: 875-881.
- Steenkamp MM, Litz BT, Hoge CW, et al. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015; 314: 489-500.
- Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psych Rev. 2003; 23: 339-376.
- Liberzon I, Abelson JL.Context processing and the neurobiology of Post-Traumatic Stress Disorder. Neuron. 2016; 92: 14-30.
- Pitman RK, Rasmusson AM, Koenen KC, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012; 13: 769-787.
- Michopoulos V, Norrholm SD, Jovanovic T. Diagnostic biomarkers for Posttraumatic Stress Disorder (PTSD): Promising horizons from translational neuroscience research. Biol Psychiat. 2015; 78: 344-353.
- James LM, Georgopoulos AP. Posttraumatic stress disorder: neuronal population coding of sudden traumatic events. Proceedings of AREADNE Research in Encoding and Decoding of Neural Ensembles, Santorini, Greece, 2018; p 67. https://areadne.org/2018/hatsopoulos-pezaris-2018-areadne.pdf
- Wang Z, Caughron B, Young MRI. Posttraumatic Stress Disorder: An immunological disorder. Front Psychiatry. 2017; 8: 222.
- Silverman MN, Pearce BD, Biron CA, et al. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005; 18: 41-78.
- Kuan PF, Waszczuk MA, Kotov R, et al. Gene expression associated with PTSD in World Trade Center responders: An RNA sequencing study. Transl Psychiatry. 2017; 7: 1297.
- Eraly SA, Nievergelt CM, Maihofer AX, et al. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry. 2014; 71: 423–431.
- Devoto C, Arcurio L, Fetta J, et al. Inflammation relates to chronic behavioral and neurological symptoms in military with traumatic brain injuries. Cell Transplant. 2016; 26: 1169–1177.
- Zohar J, Yahalom H, Kozlovsky N, et al. High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: interplay between clinical and animal studies. European Neuropsychopharmacol. 2011; 21: 796-809.
- Jones EG, Powell TP. An anatomical study of converging sensory pathways within the cerebral cortex of the monkey. Brain. 1970; 93: 793-820.
- Powell TPS. Sensory convergence in the cerebral cortex. In: Laitinen, L.V., and Livingston, K.E., eds. Surgical Approaches in Psychiatry. Baltimore, MD: University Park Press. 1973; 266-281.
- Yoshihara Y, Oka S, Nemoto Y, et al. An ICAM-related neuronal glycoprotein, telencephalin, with brain segment-specific expression. Neuron. 1994; 12: 541-553.
- Lindsberg PJ, Launes J, Tian L, et al. Release of soluble ICAM-5, a neuronal adhesion molecule, in acute encephalitis. Neurology. 2002; 58: 446-451.
- Sprenger T, Valet M, Woltmann R. Imaging pain modulation by subanesthetic S-(+)-Ketamine. Anesth Analg. 2006; 103: 729-737.
- Feder A, Parides MK, Murrough JW, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. 2014; 71: 681-688.
- Song H, Fang F, Tomasson G, et al. Association of stress-related disorders with subsequent autoimmune disease. JAMA. 2018; 319: 2388-2400.
- O'Donovan A, Cohen BE, Seal KH, et al. Elevated risk for autoimmune disorders in iraq and afghanistan veterans with posttraumatic stress disorder. Biol Psychiatry. 2015; 77: 365-374.
- Engdahl BE, James LM, Miller RD, et al. Brain Function in Gulf War Illness (GWI) and associated mental health comorbidities. J Neurol Neuromed. 2018; 3: 24-34.
- Georgopoulos AP, James LM, Mahan MY, et al. Reduced Human Leukocyte Antigen (HLA) protection in Gulf War Illness (GWI). Ebiomedicine. 3: 79-85.
- Georgopoulos AP, James LM, Carpenter AF, et al. Gulf War illness (GWI) as a neuroimmune disease. Exp Brain Res. 2017; 235: 3217-3225.
- James LM, Christova P. Engdahl BE, et al. Human leukocyte antigen (HLA) and Gulf War Illness (GWI): HLA-DRB1*13:02 spares subcortical atrophy in Gulf War veterans. Ebiomedicine. 2017; 26: 126-131.
- Neigh GN, Ali FF. Co-morbidity of PTSD and immune system dysfunction: opportunities for treatment. Curr Opin Pharmacol. 2016; 29: 104-110.
- Bettencourt A, Carvalho C, Leal B, et al. The protective role of HLA-DRB1*13 in autoimmune diseases. J Immunol Res. 2015; 15: 948723.
- Georgopoulos AP, Tsilibary EP, Souto EP, et al. Adverse effects of Gulf War Illness (GWI) serum on neural cultures and their prevention by healthy serum. J Neurol Neuromed. 2018; 3: 19-27.